Strukturuntersuchungen von Auxin-Phytohormonen. |
|
| | T1 | T2 | E [kJ/mol] |
|---|
| A | 86,54° | -95,34° |
0,000 |
|---|
| B | 106,24° | 103,40° |
0,142 |
|---|
| C | 76,23° | -31,24° |
3,003 |
|---|
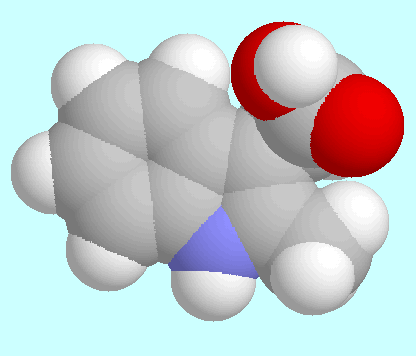
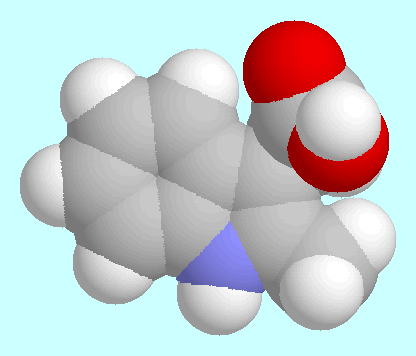
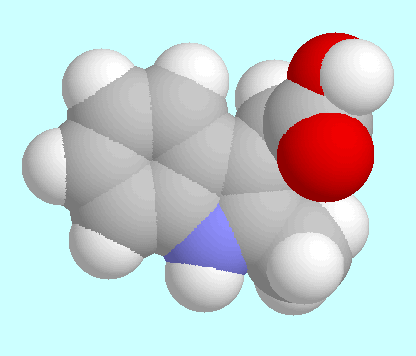
Die Potenzialfläche (PES) von
2-Methyl-3-Indolessigsäure (2-Me-IAA) ist
unten im T1/T2-Raum dargestellt.
Sie enthält drei symmetrieeindeutige lokale Minima, deren
wichtigste Daten in der Tabelle zusammengefasst sind.
Kalottenmodelle sind in energetisch aufsteigender Reihenfolge nach diesem
Absatz gezeigt.
Die lokalen Minima sind durch Reaktionswege verbunden:
zwei niederenergetische Pfade mit T1  ±100°
verbinden A--C--B--A und die Spiegelbilder
a--c--b--a,
wohingegen in den Pfaden
C--a--B und c--A--b
T1 und T2 eine gekoppelte Bewegung ausführen.
±100°
verbinden A--C--B--A und die Spiegelbilder
a--c--b--a,
wohingegen in den Pfaden
C--a--B und c--A--b
T1 und T2 eine gekoppelte Bewegung ausführen.
 |
|
Potenzialfläche von 2-Me-IAA im T1/T2-Raum.
Senkrechte Achse: T1,
horizontale Achse: T2 (jeweils von -180° bis 180°);
die Farbwechsel markieren Bereiche von
1 kcal/mol. |
Mehrere Punkte dieser PES sind bemerkenswert. Erstens ist die Struktur mit
T1=T2=0° ein Sattelpunkt zweiter Ordning, während die
analoge Struktur in
unsubstituierter IAA oder in Derivaten mit Substituenten in Position
4, 5 order 6 ein Minimum (meist sogar das globale Minimum) ist.
Zweitens hat das lokale Minimum C eine extrem geringe
Barriere in der Reaktion zu A: 0,070 kJ/mol,
das entspricht 5,85 cm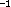 ; die entsprechende Normalschwingung von C hat eine
Frequenz von 25,5 cm; die Nullpunktsenergie dieser Schwingung liegt also über
der Potenzialbarriere. C ist deshalb ein
lokales Minimum aber kein Konformer.
; die entsprechende Normalschwingung von C hat eine
Frequenz von 25,5 cm; die Nullpunktsenergie dieser Schwingung liegt also über
der Potenzialbarriere. C ist deshalb ein
lokales Minimum aber kein Konformer.
Drittens ist die Orientierung der Methylgruppe interessant:
der Torsionswinkel T3 = H-C-C-N ist etwa 180°,
außer im Bereich um T1=0°, wo T3 = 0°
stabiler ist.
Dieses Verhalten kann durch sterische Argumente erklärt
werden: T1=0°/T3=180°
führt zu einem planaren, siebengliedrigen
Ring H-C-C-C-C-C=O, der durch eine Wasserstoffbrücke
C=O···H geschlossen wird und eine
ziemlich gespannte Struktur darstellt.
Im Gegensatz dazu ist die Anordnung mit
T1=T3=0° wesentlich entspannter; sie bildet
zwei außerhalb der Ringebene liegende
Wechselwirkungen C=O···H aus.
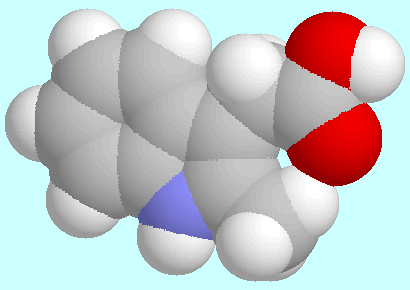
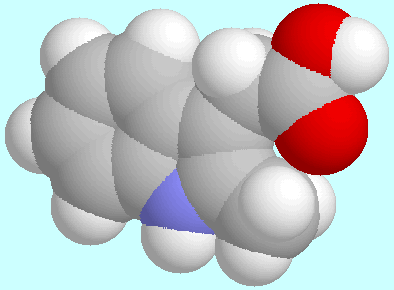
Die Energiedifferenz der beiden unten abgebildeten Strukturen ist
aber nur 1,9 kJ/mol, weil der Gewinn durch die wegfallende
Ringspannung durch die ungünstige, koplanare Anordnung
H-N-C-C-H zum Großteil kompensiert wird.

 PPAA.
PPAA.
© Publikationen
(unsere eigenen).
 Arbeitsbereich Quantenchemie.
Arbeitsbereich Quantenchemie.
Offenlegung gem. §25 MedienG: Dr. Michael Ramek, Graz.