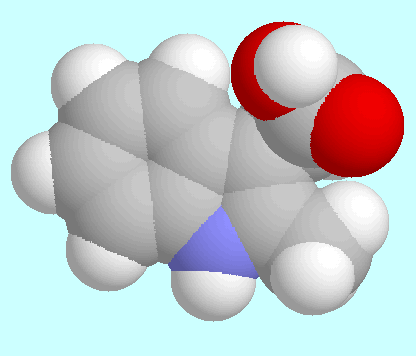
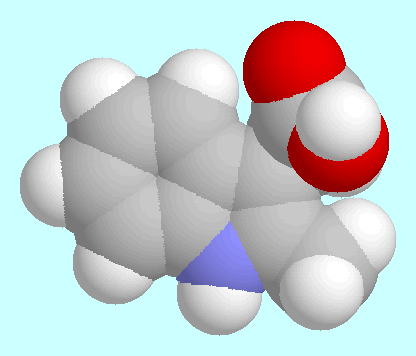
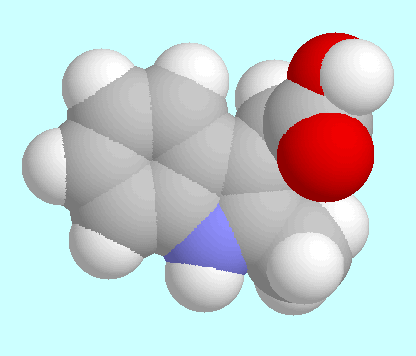
Structure Determination of Auxin Phytohormones. |
|
| | T1 | T2 | E [kJ/mol] |
|---|
| A | 86.54° | -95.34° |
0.000 |
|---|
| B | 106.24° | 103.40° |
0.142 |
|---|
| C | 76.23° | -31.24° |
3.003 |
|---|
The potential energy surface (PES) of
2-methyl-3-indole acetic acid (2-Me-IAA), which is
visualized below in T1/T2-space,
contains three symmetry-unique local minima.
Characteristic data of these minima are given in the table next to
this paragraph, fused sphere models are shown after this paragraph
in the order of their energies.
The local minima are interconnected by reaction paths:
two low energy paths with T1 approximately ±100° that
connect the local minima A--C--B--A and their mirror
images a--c--b--a
and two paths C--a--B and c--A--b,
in which T1 and T2 perform a combined motion.
 |
|
Potential energy surface of 2-Me-IAA in T1/T2-space.
Vertical axis: T1,
horizontal axis: T2 (both shown from -180° to 180°);
colour changes indicate intervals of 1 kcal/mol. |
Several points are noteworthy in this PES. The first is that
the T1=T2=0° structure is a second order saddle point;
this differs significantly from the energy surfaces
of unsubstituted IAA or derivatives with
substituents at positions 4, 5, and 6, because
in these compounds the T1=T2=0° structure is a local minimum,
in most cases even the global minimum.
Secondly, the local minimum C has an extremely
low barrier in the reaction to A: 0.070 kJ/mol,
equivalent to 5.85 cm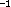 ; the corresponding normal mode of C has a
frequency of 25.5 cm, which means that the zero point energy of that motion is
higher than the potential energy barrier. C therefore
is a local minimum but no stable conformer.
; the corresponding normal mode of C has a
frequency of 25.5 cm, which means that the zero point energy of that motion is
higher than the potential energy barrier. C therefore
is a local minimum but no stable conformer.
The third point concerns
the orientation of the methyl group,
which is described by a torsion angle T3 = H-C-C-N close to 180°,
except for the region around T1=0°, where
T3 close to 0° is more stable.
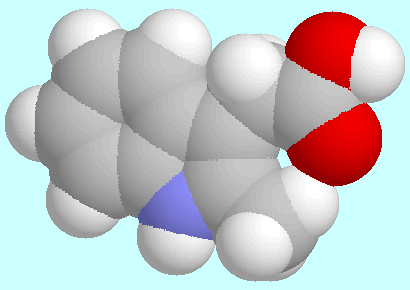
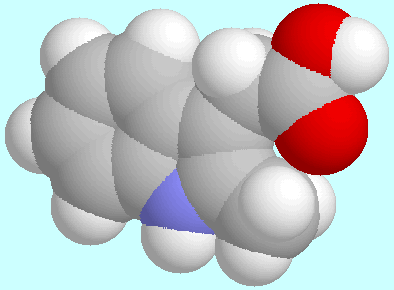
This can be explained by sterical reasons: T1=0°/T3=180°
leads to the formation of a planar,
seven-membered ring H-C-C-C-C-C=O that
is closed by a hydrogen bond C=O···H.
This ring, which is shown in the display below this paragraph,
is of considerable strain, whereas the
T1=T3=0° orientation, which also shown below, leads to an arrangement
with two out-of-plane C=O···H interactions.
Interestingly, the T3=0° form is only 1.9 kJ/mol lower
in energy than the T3=180° form, because the gain in sterical
energy is cancelled by the unfavourable, co-planar H-N-C-C-H orientation.

 PPAA.
PPAA.
© publications
(our own).
 Quantum Chemistry Group.
Quantum Chemistry Group.
Informations required by Austrian law
(Offenlegung gem. §25 MedienG): Dr. Michael Ramek, Graz.