Dr. Michael Ramek: Forschung.
 English version.
English version.Dr. Michael Ramek: Forschung. |
English version. |
Hauptforschungsgebiet ist die Strukturbestimmung von biologisch
wichtigen Verbindungen mittels quantenchemischer ab initio Methoden.
 Für mehrere Jahre lag der Schwerpunkt auf der Untersuchung von
intramolekularen Wasserstoffbrückenbindungen
in Amino- und Hydroxyverbindungen (Projekte
P6856 (1988-1990),
P8053 (1990-1991) und
P9095 (1992-1994)
des FWF); die Ergebnisse dieser Arbeiten haben z.B. in Zusammenarbeit mit
Prof. Paul G. Mezey (University of Saskatchewan, Saskatoon,
Kanada) Anwendung auf dem Gebiet der Form-Ähnlichkeits-Analyse
gefunden. In den letzten Jahren standen Derivate von
Indol-3-Essigsäure und Modellpeptide
im Vordergrund des Interesses (siehe unten).
Für mehrere Jahre lag der Schwerpunkt auf der Untersuchung von
intramolekularen Wasserstoffbrückenbindungen
in Amino- und Hydroxyverbindungen (Projekte
P6856 (1988-1990),
P8053 (1990-1991) und
P9095 (1992-1994)
des FWF); die Ergebnisse dieser Arbeiten haben z.B. in Zusammenarbeit mit
Prof. Paul G. Mezey (University of Saskatchewan, Saskatoon,
Kanada) Anwendung auf dem Gebiet der Form-Ähnlichkeits-Analyse
gefunden. In den letzten Jahren standen Derivate von
Indol-3-Essigsäure und Modellpeptide
im Vordergrund des Interesses (siehe unten).
 Ein vollkommen anderes, nunmehr weitgehend abgeschlossenes Thema ist
die Symmetrisierung
von Wellenfunktionen und Quantenzuständen, die in
einer Reihe von Publikationen,
zum Großteil in Zusammenarbeit mit Prof. Bruno Gruber
(Southern Illinois University, Carbondale, IL (USA)),
untersucht wurde.
Ein vollkommen anderes, nunmehr weitgehend abgeschlossenes Thema ist
die Symmetrisierung
von Wellenfunktionen und Quantenzuständen, die in
einer Reihe von Publikationen,
zum Großteil in Zusammenarbeit mit Prof. Bruno Gruber
(Southern Illinois University, Carbondale, IL (USA)),
untersucht wurde.
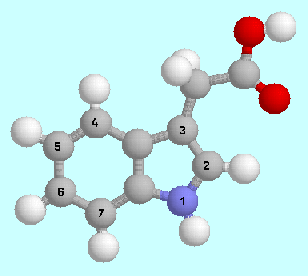 Indol-3-Essigsäure
und Derivate dieser Verbindung sind Wachstumshormone
in Pflanzen (Auxine), die unter anderem Zellteilung,
die Wachstumsentwicklung sowie die Synthese spezifischer Proteine steuern.
In einer
Reihe von Arbeiten, die zusammen mit
Dr. Sanja Tomić (Institut Ruđer Bošković, Zagreb,
Kroatien)
entstanden sind, konnte gezeigt werden, dass die Energiehyperflächen
dieser Verbindungen entscheidend von der Substitution an einer bestimmten
Position am Indolkern (Position 4) abhängen, während die
Substitution durch Chlor oder eine Ethylgruppe an anderen Positionen
des Phenylringes kaum einen Einfluss hat. Obwohl die Form der
Potenzialfläche nicht direkt mit der biologischen Aktivität
korreliert werden kann, da diese auch von anderen Parametern
abhängt (wie der Lipophilizität oder
dem Einfluss anderer Verbindungen, wie z.B. Cytokinine),
so geben diese Studien doch wichtige Erkenntnisse hinsichtlich der
Faktoren, die für die Auxinaktivität entscheidend sind.
Indol-3-Essigsäure
und Derivate dieser Verbindung sind Wachstumshormone
in Pflanzen (Auxine), die unter anderem Zellteilung,
die Wachstumsentwicklung sowie die Synthese spezifischer Proteine steuern.
In einer
Reihe von Arbeiten, die zusammen mit
Dr. Sanja Tomić (Institut Ruđer Bošković, Zagreb,
Kroatien)
entstanden sind, konnte gezeigt werden, dass die Energiehyperflächen
dieser Verbindungen entscheidend von der Substitution an einer bestimmten
Position am Indolkern (Position 4) abhängen, während die
Substitution durch Chlor oder eine Ethylgruppe an anderen Positionen
des Phenylringes kaum einen Einfluss hat. Obwohl die Form der
Potenzialfläche nicht direkt mit der biologischen Aktivität
korreliert werden kann, da diese auch von anderen Parametern
abhängt (wie der Lipophilizität oder
dem Einfluss anderer Verbindungen, wie z.B. Cytokinine),
so geben diese Studien doch wichtige Erkenntnisse hinsichtlich der
Faktoren, die für die Auxinaktivität entscheidend sind.
 Peptide
werden meist durch Anfügen einer Formyl- oder Acetylgruppe
an den N-Terminus einer kurzen Peptidkette und Konversion
des C-Terminus in eine Amidgruppe modelliert.
Die Peptidkette kann im Extremfall aus einem einzigen Aminosäurerest
X bestehen; in diesem Fall enthält die Modellverbindung
Peptide
werden meist durch Anfügen einer Formyl- oder Acetylgruppe
an den N-Terminus einer kurzen Peptidkette und Konversion
des C-Terminus in eine Amidgruppe modelliert.
Die Peptidkette kann im Extremfall aus einem einzigen Aminosäurerest
X bestehen; in diesem Fall enthält die Modellverbindung